Robots are not just limited to the assembly line. At NVIDIA, Liila Torabi works on making the next generation of robotics possible. Torabi is the senior product manager for Isaac Sim, a robotics and AI simulation platform powered by NVIDIA Omniverse.
Torabi spoke with NVIDIA AI Podcast host Noah Kravitz about the new era of robotics, one driven by making robots smarter through AI.
Isaac Sim is used to power photorealistic, physically accurate virtual environments to develop, test and manage AI-based robots.
Key Points From This Episode:
To get to a point where robots and humans can interact and work together, developers need to train the robots and simulate their behavior ahead of time to ensure performance, safety and a variety of other factors. This is where Isaac Sim comes into play.
For Torabi, the biggest technical hurdle is having the robot do more sophisticated jobs. Robot manipulation with different objects, shapes and environments is a challenge.
Tweetables:
“For robotics to get into the next era, we need it to be smarter, so we need the AI component to this.” — Liila Torabi [8:08]
“NVIDIA is well positioned for playing an important role in this next era of robotics because not only do we have the hardware for it, we know how to use this hardware to make this thing smarter. That’s why I’m very excited to see where we can go with Isaac Sim. ” — Liila Torabi [12:09]
Robots can do amazing things. Compare even the most advanced robots to a three-year-old, however, and they can come up short. UC Berkeley Professor Pieter Abbeel has pioneered the idea that deep learning could be the key to bridging that gap: creating robots that can learn how to move through the world more fluidly and naturally.
What if robots could learn, teach themselves and pass on their knowledge to other robots? Where could that take machines and the notion of machine intelligence? And how fast could we get there? Sergey Levine, an assistant professor at UC Berkeley’s department of Electrical Engineering and Computer Sciences, explores these questions and more.
NVIDIA’s Jetson interns, recruited at top robotics competitions, discuss what they’re building with NVIDIA Jetson, including a delivery robot, a trash-disposing robot and a remote control car to aid in rescue missions.
Self-driving startup AutoX last week took the wraps off its “Gen5” self-driving system. The autonomous driving platform, which is specifically designed for robotaxis, uses NVIDIA DRIVE automotive-grade GPUs to reach up to 2,200 trillion operations per second (TOPS) of AI compute performance.
In January, AutoX launched a commercial robotaxi system in Shenzhen, China, becoming one of the first autonomous driving companies in the world to provide full self-driving mobility services with no safety driver behind the wheel. The Gen5 system is the next step in its global rollout of safer, more efficient autonomous transportation.
“Safety is key. We need higher processing performance for safe and scalable robotaxi operations,” said Jianxiong Xiao, founder and CEO at AutoX. “With NVIDIA DRIVE, we now have power for more redundancy in a form factor that is automotive grade and more compact.”
Zero Blind Spots
In developing safe self-driving technology, AutoX is aimed at solving the toughest environments first — specifically high-traffic, urban areas.
At the Gen5 Release Event, the company livestreamed its fully driverless robotaxi transporting a passenger through challenging narrow streets in China, called the “Urban Village.”
Safely navigating such chaotic streets requires sensors that can detect obstacles and other road users with the highest levels of accuracy. The Gen5 system relies on 28 automotive-grade camera sensors generating more than 200 million pixels per frame 360-degrees around the car. (For comparison, a single high-definition video frame contains about 2 million pixels.)
In addition to cameras, the robotaxi system includes six high-resolution lidar sensors that produce 15 million points per second and surround 4D radar.
At the center of the Gen5 system are two NVIDIA Ampere architecture GPUs that deliver 900 TOPS each for a truly level 4 autonomous, production platform. With this unprecedented level of AI compute at the core, Gen5 has enough performance to power ultra complex self-driving DNNs while maintaining the compute headroom for more advanced upgrades.
This capability makes it possible for the vehicles to react to high-traffic situations — like dozens of motorcycles and scooters cutting in or riding the opposite way at the same time — in real time, and continually improving, learning how to manage new scenarios as they arise.
More Stops Added
The Shenzhen fully driverless robotaxi service is just the first stop in AutoX’s roadmap to deploy a global driverless vehicle platform.
With a population of more than 12 million people and ranking in the top 50 of global cities with the heaviest traffic, Shenzhen provides an ideal setting for developing a scalable robotaxi model.
The startup plans to roll out thousands of autonomous vehicles powered by the Gen5 system over the next couple of years and expand to multiple cities around the world. AutoX is working with partners such as Stellantis and Honda to integrate their technology in a variety of vehicle platforms.
By leveraging the open, scalable NVIDIA DRIVE platform for each of these use cases, the opportunities for the road ahead are limitless.
Highlighting deep support from a flourishing roster of GeForce partners, NVIDIA’s Jeff Fisher delivered a virtual keynote at COMPUTEX 2021 in Taipei Tuesday.
The virtual keynote, which led with Fisher talking about gaming and then NVIDIA’s Manuvir Das, head of enterprise computing, talking about AI and enterprise platforms (see wrapup, here), began by highlighting NVIDIA’s deep ties to Taiwan.
Deep Roots in Taiwan
Fisher announced the release of a mod — one of thousands for the hyperrealistic flight simulator — paying tribute to Taipei.
“We miss Taipei and wish we could be there in person for COMPUTEX,” Fisher said. “So we created Taipei City in Microsoft Flight Sim and flew in virtually on a GeForce RTX 3080.”
The callout was a tribute to NVIDIA’s many close partners in Taiwan, including Acer, AOC, ASUS, GIGABYTE, MSI and Palit.
COMPUTEX is also a key gathering point for partners from around the world, including Alienware, Colorful, Dell, EVGA, Gainward, Galax, HP, Inno3D, Lenovo, PNY, Razer, ViewSonic and Zotac.
“It’s always great to talk directly to our partners, and this year we have a lot to talk about,” Fisher said.
GeForce Partners in Every Category
Throughout his talk, Fisher highlighted NVIDIA’s close ties to partners in Taiwan — and throughout the world — in gaming laptops, desktop GPUs, studio laptops and G-SYNC displays.
Thanks to decades of work with partners, gaming laptops are thriving, and Fisher spotlighted six GeForce RTX laptops from Acer, Dell and HP.
This year brought a record launch for RTX laptops, with over 140 models from every manufacturer.
Starting at $799 and featuring Max-Q, a collection of NVIDIA technologies for making gaming laptops thinner, lighter and more powerful, “there is now an RTX laptop for every gamer,” Fisher said.
Highlighting one example, Fisher announced the Alienware x15, an ultra-thin, GeForce RTX 3080 laptop.
Powered by Max-Q technologies including Dynamic Boost 2.0, WhisperMode 2.0 and Advanced Optimus, and featuring a 1440p display, “it is the world’s most powerful sub-16mm 15-inch gaming laptop,” Fisher said.
In the desktop category, the RTX family of desktop GPUs gets a new flagship gaming GPU, the GeForce RTX 3080 Ti, and the GeForce RTX 3070 Ti.
NVIDIA partners announced 98 new desktop GPU products, with 11 key partners announcing new RTX 3080 Ti and 3070 Ti desktop graphics cards.
With second-generation RT Cores and third-generation Tensor Cores, the NVIDIA Ampere architecture is “our greatest generational leap ever,” Fisher said. “The 80 Ti class of GPUs represents the best of our gaming lineup.”
For 3D designers, video editors and photographers, NVIDIA developed NVIDIA Studio. These are specially configured systems, optimized and tested for creator workflows, and supported with a monthly cadence of Studio drivers, Fisher said.
NVIDIA partners announced 8 new Studio products, including six ConceptD laptops from Acer and two laptops from HP.
The 14-inch HP Envy brings the capabilities of RTX to an ultra-portable laptop that’s “great for students and creators on the go,” Fisher said.
The new Acer ConceptD offers a variety of traditional clamshell options and an Ezel sketch board design to give creators even more flexibility, Fisher said.
In displays, NVIDIA partners announced five new G-SYNC products. They included two G-SYNC ULTIMATE displays and three G-SYNC displays from Acer, MSI and ViewSonic.
“G-SYNC introduced stutter-free gaming,” Fisher said. “With over 20 trillion buttery-smooth pixels now shipped once you game with G-SYNC, there is no turning back.”
The spate of announcements — highlighted in Fisher’s keynote — are being celebrated throughout the week at COMPUTEX.
Acer, Alienware, and MSI all had special digital activations to support their new products.
“Thanks to all our partners who are just as excited as we are about reinventing this market, and are joining us in the next major leap forward,” Fisher said.
Microsoft Azure has announced the general availability of the ND A100 v4 VM series, their most powerful virtual machines for supercomputer-class AI and HPC workloads, powered by NVIDIA A100 Tensor Core GPUs and NVIDIA HDR InfiniBand.
When solving grand challenges in AI and HPC, scale is everything. Natural language processing, recommendation systems, healthcare research, drug discovery and energy, among other areas, have all seen tremendous progress enabled by accelerated computing.
Much of that progress has come from applications operating at massive scale. To accelerate this trend, applications need to run on architecture that is flexible, accessible and can both scale up and scale out.
The ND A100 v4 VM brings together eight NVIDIA A100 GPUs in a single VM with the NVIDIA HDR InfiniBand that enables 200Gb/s data bandwidth per GPU. That’s a massive 1.6 Tb/s of interconnect bandwidth per VM.
And, for the most demanding AI and HPC workloads, these can be further scaled out to thousands of NVIDIA A100 GPUs under the same low-latency InfiniBand fabric, delivering both the compute and networking capabilities for multi-node distributed computing.
Ready for Developers
Developers have multiple options to get the most performance out of the NVIDIA A100 GPUs in the ND A100 v4 VM for their applications, both for application development and managing infrastructure once those applications are deployed.
To simplify and speed up development, the NVIDIA NGC catalog offers ready-to-use GPU-optimized application frameworks, containers, pre-trained models, libraries, SDKs and Helm charts. With the prebuilt NVIDIA GPU-optimized Image for AI and HPC on the Azure Marketplace, developers can get started with GPU-accelerated software from the NGC catalog with just a few clicks.
The ND A100 v4 VMs are also supported in the Azure Machine Learning service for interactive AI development, distributed training, batch inferencing and automation with ML Ops.
Deploying machine learning pipelines in production with ND A100 v4 VMs is further simplified using the NVIDIA Triton Inference Server, an open-source inference serving application that’s integrated with Azure ML to maximize both GPU and CPU performance and utilization to help minimize the operational costs of deployment.
Developers and infrastructure managers will soon also be able to use Azure Kubernetes Service, a fully managed Kubernetes service to deploy and manage containerized applications on the ND A100 v4 VMs, with NVIDIA A100 GPUs.
Learn more about the ND A100 v4 VMs on Microsoft Azure and get started with building innovative solutions on the cloud.
The streaming device with the most 4K HDR content available now has even more as Apple TV joins the lineup on NVIDIA SHIELD.
Starting today, SHIELD owners can stream the Apple TV app, including Apple TV+, and its impressive lineup of award-winning series, compelling dramas, groundbreaking documentaries, kids’ shows, comedies and more.
The Apple TV app features Apple TV+, Apple’s video subscription service featuring Apple Originals, including series like Ted Lasso, The Morning Show, For All Mankind and Servant; as well as movies like Greyhound, Palmer and Wolfwalkers. With SHIELD TV, they can be enjoyed in stunning Dolby Vision — a combination that offers beautiful 4K HDR picture quality with remarkable colors, sharper contrasts and incredible brightness. Dolby Atmos is also supported, unlocking richer and more immersive sound. Together, they deliver the best possible cinematic experience.
Apple TV channels are also on the Apple TV app, such as AMC+, Paramount+ and Starz, and watch ad-free and on demand, directly on the Apple TV app. Through Family Sharing, up to six family members can share subscriptions to Apple TV channels using their personal Apple ID and password. And you can enjoy personalized and curated recommendations and access your library of movies and shows purchased from Apple.
Apple TV also works with the built-in Google Assistant on SHIELD for hands-free control of your entertainment. Use simple voice commands to pause, rewind, fast-forward and more.
To search for content using voice, say “Hey Google,” or press the microphone button, and call out a genre or specific show you’d like to watch. New Apple Originals arrive every month, so there’s always something binge-worthy to watch.
And with SHIELD’s amazing AI-upscaling technology even HD content gets an eye-popping boost to 4K.
Download Apple TV from the Google Play store. There’s even a free seven-day Apple TV+ trial available for new subscribers.
SHIELD Gets Even Better
SHIELD is the faster, smarter streaming media player. It’s built on Android TV and supported by industry-leading software updates, includes the latest audio and visual technology and offers the most 4K HDR movies and shows.
With HBO Max delivering new 4K HDR Dolby Vision and Dolby Atmos cinematic releases every month and YouTube TV adding more channels and live sports from Hulu, there’s never been a better time to pick up a SHIELD TV.
This includes those who like to game before they watch: For a limited time, get a three-month GeForce NOW Priority Membership with the purchase of a SHIELD TV. GeForce NOW instantly transforms SHIELD TVs — plus PCs, Macs, Chromebooks or mobile devices — into the PC gaming rigs gamers have long dreamed of.
Fisher, senior vice president of NVIDIA’s GeForce business, announced a pair of powerful new gaming GPUs — the GeForce RTX 3080 Ti and GeForce 3070 Ti. He spoke about NVIDIA’s role in powerful new laptops for gamers and creators. And he detailed the fast-growing adoption of NVIDIA RTX technologies in a growing roster of games.
Das, head of enterprise computing, announced that dozens of new servers are certified to run NVIDIA AI Enterprise software, marking a rapid expansion of the NVIDIA-Certified Systems program, which has grown to include more than 50 systems from the world’s leading manufacturers.
The joint keynote began with a flourish, with Fisher announcing a unique Microsoft Flight Simulator mod paying tribute to Taipei. “We miss Taipei and wish we could be there in person for COMPUTEX,” Fisher said. “So we created Taipei City in Microsoft Flight Sim and flew in virtually on a GeForce RTX 3080.”
NVIDIA’s Jeff Fisher announced a unique Microsoft Flight Simulator mod paying tribute to Taipei.
New Gaming Flagship, New Laptops, RTX Momentum
Fisher began the talk by touching on how gaming has transformed entertainment. NVIDIA RTX technologies are changing everything, not just for gamers, but for 150 million creators, broadcasters and students, Fisher explained.
RTX accelerates the No. 1 photography app, the No. 1 video editing app and the No. 1 broadcast app. For gamers, RTX is powering the No. 1 battle royale, the No. 1 RPG and the No. 1 best-selling game of all time. Soon 12 of the top 15 competitive shooters will feature NVIDIA Reflex, with the addition of Escape from Tarkov, CrossFire HD and War Thunder, Fisher said.
And GeForce laptops are the fastest-growing gaming platform for such experiences. It’s a platform now fueled top to bottom by RTX, third-generation Max-Q technologies and the magic of DLSS, which uses AI-powered Tensor Cores to boost frame rates.
This year brought a record launch for RTX laptops, with over 140 models from every manufacturer. Starting at $799 and featuring Max-Q, a collection of NVIDIA technologies for making gaming laptops thinner, lighter and more powerful, “there is now an RTX laptop for every gamer,” Fisher said.
Highlighting an example of that work, Fisher announced the Alienware x15, an ultra-thin, GeForce RTX 3080 laptop. Powered by Max-Q technologies including Dynamic Boost 2.0, WhisperMode 2.0 and Advanced Optimus, and featuring a 1440p display, “it is the world’s most powerful sub-16mm 15-inch gaming laptop,” Fisher said.
For 3D designers, video editors and photographers, NVIDIA developed NVIDIA Studio. These are specially configured systems, optimized and tested for creator workflows, and supported with a monthly cadence of Studio Drivers, Fisher said.
Fisher announced a pair of new Studio laptops from HP and Acer. The 14-inch HP Envy brings the capabilities of RTX to an ultra-portable laptop that’s “great for students and creators on the go,” Fisher said. The new Acer ConceptD offers a variety of traditional clamshell options and an Ezel sketch board design to give creators even more flexibility, Fisher said.
The RTX family gets a new flagship gaming GPU, the GeForce RTX 3080 Ti.
Based on the NVIDIA Ampere architecture, with second-generation RT Cores and third-generation Tensor Cores, Ampere is “our greatest generational leap ever,” Fisher said. “The 80 Ti class of GPUs represents the best of our gaming lineup.”
The GPU comes as the production value of games continues to march forward, Fisher explained. “New titles like Cyberpunk 2077 and Watch Dogs: Legion have elevated realism, demanding even more of the GPU,” he said.
Fisher said the RTX 3080 Ti is 1.5x faster than its predecessor and tears through the latest games with all the settings cranked up. And the RTX 3070 Ti is 1.5x faster than a 2070 SUPER, thanks to more cores and superfast GDDR6X memory.
GeForce RTX 3080 Ti availability will begin on June 3, starting at $1,199. GeForce RTX 3070 Ti availability begins on June 10, starting at $599.
“Every person born today is a gamer,” Fisher said. “We have an amazing future ahead, and we look forward to building it with all of you.”
‘AI for Every Company’
Following Fisher, Das spoke in detail about the three essential ingredients built by NVIDIA: the hardware foundation from which to make any system, the software platform for artificial intelligence and the software platform for collaborative design.
“It is time to democratize AI by bringing its transformative power to every company and its customers,” Das said.
To help system manufacturers create AI-optimized designs and to ensure that the systems can be relied on by customers, NVIDIA made NVIDIA-Certified, a program for servers that incorporate GPU acceleration, Das explained.
NVIDIA’s Manuvir Das spoke in detail about the three essential ingredients built by NVIDIA: the hardware foundation from which to make any system, the software platform for artificial intelligence and the software platform for collaborative design.
Coming from Advantech, Altos, ASRock Rack, ASUS, Dell Technologies, GIGABYTE, Hewlett Packard Enterprise, Lenovo, QCT, Supermicro and others, the growing roster of NVIDIA-Certified Systems include some of the highest-volume x86 servers used in mainstream data centers. They bring the power of AI to a wide range of industries including healthcare, manufacturing, retail and financial services.
“Going forward, the DPU will be an essential component of every server, in the data center and at the edge,” Das said.
Das also announced the NVIDIA-Certified System program will expand to support accelerated systems with Arm-based host CPUs. In addition, along with NVIDIA partner GIGABYTE, Das announced a devkit can be used by application developers to prepare their GPU-accelerated apps for Arm.
“As the GPU and DPU accelerators take on more of the compute workload for AI, it becomes useful to view the host CPU as an orchestrator, more so than as the compute engine,” Das said.
Google Cloud is among the first cloud service providers planning to enable NVIDIA’s powerful Base Command Platform for the management and orchestration of clusters in their cloud instances.
Building on NVIDIA’s work to enable powerful GPU instances in the cloud, Das announced that Google Cloud is among the first cloud service providers planning to enable NVIDIA’s powerful Base Command Platform for the management and orchestration of clusters in their cloud instances.
The software is designed for large-scale, multi-user and multi-team AI development workflows hosted either on premises or in the cloud. It enables numerous researchers and data scientists to simultaneously work on accelerated computing resources, helping enterprises maximize the productivity of both their expert developers and their valuable AI infrastructure.
Das announced that Cloudera, a provider of Apache Spark to enterprise data centers around the world, will add transparent GPU acceleration using NVIDIA RAPIDS, a set of libraries that accelerate machine learning on GPUs. A fully integrated solution from Cloudera will be available starting this summer, with the release of CDP version 7.1.7, Das said.
Das also announced that NVIDIA will partner with leading global systems providers to offer NVIDIA Omniverse Enterprise, making it easy for teams to collaboratively design and simulate in 3D at $14,000 per year per company.
“Companies around the world and across industries are already using Omniverse to collaborate in amazing ways,” Das said.
And in security, Das announced that NVIDIA is working with Red Hat to provide Morpheus developer kits for both OpenShift and Red Hat Enterprise Linux, or RHEL, the most commonly used version of commercial Linux in enterprise data centers today.
Morpheus, introduced at NVIDIA’s GTC conference earlier this year, uses machine learning to identify, capture and take action on threats and anomalies.
“Cybersecurity companies will now be able to use Morpheus on RHEL and OpenShift to bring advanced security to every enterprise data center,” Das said.
‘Thank You’
Das closed his COMPUTEX talk — which touched on the work of so many partners — on a note of gratitude to NVIDIA’s customers and partners throughout the industry.
“On behalf of my co-presenter Jeff Fisher, our CEO Jensen Huang and all of NVIDIA, thank you,” Das said. “Thank you for joining us today; thank you for being with us every step of the way; and thank you for continuing forward with us on this amazing journey.”
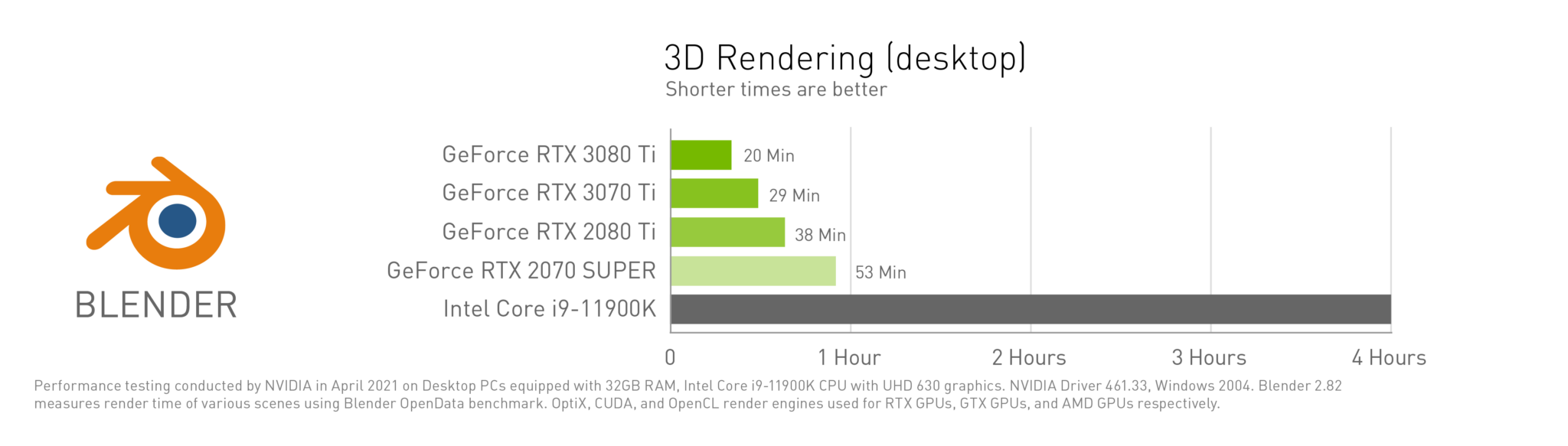
Content creators are getting a slew of new tools in their creative arsenal at COMPUTEX with the announcement of new NVIDIA GeForce RTX 3080 Ti and 3070 Ti GPUs. These GPUs deliver massive time savings for freelancers and creatives specializing in video editing, 3D animation or architectural visualization.
In addition, HP and Acer announced new NVIDIA Studio laptops with GeForce RTX 30 Series and NVIDIA RTX professional GPUs, including the latest 11th Gen Intel mobile processors.
Finally, the latest NVIDIA Omniverse app, Machinima, is now in beta, fully supported by the latest NVIDIA Studio drivers.
Expand Your Creative Arsenal
The GeForce RTX 3080 Ti and 3070 Ti GPUs deliver incredible leaps in performance and fidelity. The powerhouse 3080 Ti is ideal for high-end content creation thanks to its faster clocks and 12GB of superfast GDDR6X video memory.
The 3080 Ti shines in heavy-rendering workloads, where it can make full use of the NVIDIA Ampere architecture, including second-generation RT Cores and third generation Tensor Cores, as well as performance gains from NVIDIA DLSS, to get the job done fast. And it’s ideal for high-end video editing, especially 8K HDR RAW footage.
The GeForce RTX 3070 Ti turbocharges the popular RTX 3070 by giving it more CUDA cores and super-speedy GDDR6 video memory. Content creators will enjoy faster texture loading times, making the 3070 Ti a great choice for 3D rendering and video editing with up to 6K HDR RAW footage.
The GeForce RTX 3080 Ti will be available on June 3 starting at $1,199, and the GeForce RTX 3070 Ti will be available next week starting at $599.
More Studio Laptop Options for Content Creation
HP, Acer and MSI are offering new NVIDIA Studio laptops powered by the latest GeForce RTX 30 Series and NVIDIA RTX Professional GPUs, and the latest 11th Gen Intel mobile processors.
HP’s refreshed Envy 15 and Envy 14 feature elegant designs and fantastic, color-calibrated displays. For students and commuters, the Envy 14 packs the power of NVIDIA Studio in an ultra-portable creator laptop, powered with a GeForce RTX 3050 or 3050 Ti GPU. For creators who want more power, the Envy 15, which can include up to a GeForce RTX 3060 Laptop GPU, is perfect for video editing thanks to its larger display and video memory.
Acer is updating the ConceptD creator lineup with the new ConceptD 3 Ezel, the all-metal ConceptD 5 laptop and the ConceptD 7 Ezel flagship. The ConceptD 7 Ezel features up to a GeForce RTX 3080 and the Pro model up to an NVIDIA RTX A5000 Laptop GPU, and Acer’s patented adjustable 15.6-inch 4K PANTONE-validated touchscreen display. The lineup has a wide selection of GPU options for any type of creator, ranging from the GeForce RTX 3050 Ti up to RTX 3080, and with options for professional GPUs such as the NVIDIA RTX A5000.
MSI’s Creator Z16, Creator 17, WS66 and WS76 units have joined the NVIDIA Studio program. The Creator Z16 is an elegant and premium laptop powered by a GeForce RTX 3060 Laptop GPU. It sports a striking postmodern design with a True Pixel display with QHD+ resolution, DCI-P3 100 percent color gamut, factory-calibrated Delta-E <2 out of the box accuracy.
Omniverse Machinima Now in Beta
Gaming has been an inspiration to digital creators and artists for decades. Machinima, an art form started in the ’90s, uses real-time 3D technologies with game assets, visual effects and character animation to create short, humorous clips, or full-length movies. Using the latest RTX technologies, NVIDIA built Omniverse Machinima, an application that lets people collaborate in real time and create amazing animated video game stories.
Creators can import their own game assets or draw from Omniverse’s growing library, including content from Mount & Blade II: Bannerlord, Squad! and iconic NVIDIA tech demos. Advanced physics effects like destruction or fire can be applied with NVIDIA’s PhysX 5, Blast and Flow. Animate character movements using only a webcam with wrnch’s AI Pose and use simple audio tracks to animate character faces with Omniverse Audio2Face.
Upon completion, creators can access the Omniverse RTX Renderer for the highest quality output, enhanced with real-time ray tracing and referenced path tracing for interactive, bleeding-edge visuals.
These new GPUs and NVIDIA Studio laptops benefit from the RTX acceleration and optimization of the NVIDIA Studio ecosystem:
Performance optimization in hundreds of creator apps, including 70+ apps optimized for RTX ray tracing and NVIDIA DLSS.
NVIDIA Studio Drivers, optimized for content creation to provide the best performance and stability. Drivers are kept up to date automatically with GeForce Experience and NVIDIA RTX Experience.
Several of the world’s top server manufacturers today at COMPUTEX 2021 announced new systems powered by NVIDIA BlueField-2 data processing units. While systems with NVIDIA GPUs are already available now with the option of adding BlueField-2 DPUs, many applications and customer use cases may not require a GPU but can still benefit from a DPU.
The new servers from ASUS, Dell Technologies, GIGABYTE, QCT and Supermicro are ideal for enterprises seeking the additional performance, security and manageability that NVIDIA BlueField-2 DPUs offer.
The NVIDIA BlueField-2 DPU is the world’s leading data processing unit.
Servers that primarily run software-defined networking (for example, a stateful load balancer or distributed firewall), software-defined storage or traditional enterprise applications will all benefit from the DPU’s ability to accelerate, offload and isolate infrastructure workloads for networking, security and storage.
Systems running VMware vSphere, Windows or hyperconverged infrastructure solutions also benefit from including a DPU, whether running AI and machine learning applications, graphics-intensive workloads or traditional business applications.
NVIDIA BlueField DPUs shift infrastructure tasks from the CPU to the DPU, making more server CPU cores available to run applications, which increases server and data center efficiency. The DPU places a “computer in front of the computer” for each server, delivering separate, secure infrastructure provisioning that is isolated from the server’s application domain. This allows agentless workload isolation, security isolation, storage virtualization, remote management and telemetry on both virtualized and bare-metal servers.
Asus, Dell Technologies, GIGABYTE, QCT and Supermicro have announced plans to offer servers accelerated by NVIDIA BlueField-2 DPUs.
Customers and software makers can program BlueField DPUs easily using the NVIDIA DOCA SDK, the data-center-on-a-chip architecture that simplifies application development and ensures forward and backward compatibility with the NVIDIA BlueField-3 DPU, expected in 2022, and all future BlueField DPUs.
Enterprise customers are interested in deploying DPUs in many or even all of their data center servers. The availability of DPU-accelerated servers from leading system builders makes it simple and easy for customers to buy the market-leading BlueField-2 DPU pre-installed and pre-configured with their favorite servers.
BlueField-2 DPU-accelerated servers are expected this year, with several anticipated to submit to the NVIDIA-Certified Systems program when DPU server certification becomes available.
What’s a three-letter acronym for a “video-handling chip”? A GPU, of course. Who knew, though, that these parallel processing powerhouses could have a way with words, too.
Following a long string of victories for computers in other games — chess in 1997, go in 2016 and Texas hold’em poker in 2019 — a GPU-powered AI has beaten some of the world’s most competitive word nerds at the crossword puzzles that are a staple of every Sunday paper.
Dr.Fill, the crossword puzzle-playing AI created by Matt Ginsberg — a serial entrepreneur, pioneering AI researcher and former research professor — scored higher than any humans last month at the American Crossword Puzzle Tournament.
Dr.Fill’s performance against more than 1,300 crossword enthusiasts comes after a decade of playing alongside humans through the annual tournament.
Such games, played competitively, test the limits of how computers think and better understand how people do, Ginsberg explains. “Games are an amazing environment,” he says.
Dr.Fill’s edge? A sophisticated neural network developed by UC Berkeley’s Natural Language Processing team — trained in just days on an NVIDIA DGX-1 system and deployed on a PC equipped with a pair of NVIDIA GeForce RTX 2080 Ti GPUs — that snapped right into the system Ginsberg had been refining for years.
A way with words: A pair of NVIDIA RTX 2080 Tis powered a sophisticated neural network developed by UC Berkeley’s Natural Language Processing team.
“Crossword fills require you to make these creative multi-hop lateral connections with language,” says Professor Dan Klein, who leads the Natural Language Processing team. “I thought it would be a good test to see how the technology we’ve created in this field would handle that kind of creative language use.”
Given that unstructured nature, it’s amazing that a computer can compete at all. And to be sure, Dr.Fill still isn’t necessarily the best, and that’s not only because the American Crossword Puzzle Tournament’s official championship is reserved only for humans.
The contest’s organizer, New York Times Puzzle Editor Will Shortz, pointed out that Dr.Fill’s biggest advantage is speed: it can fill in answers in an instant that humans have to type out. Judged solely by accuracy, however, Dr.Fill still isn’t the best, making three errors during the contest, worse than several human contestants.
Nevertheless, Dr.Fill’s performance in a challenge that, unlike more structured games such as chess or go, rely so heavily on real-world knowledge and wordplay is remarkable, Shortz concedes.
“It’s just amazing they have programmed a computer to solve crosswords — especially some of the tricky hard ones,” Shortz said.
A Way with Words
Ginsberg, who holds a Ph.D. in mathematics from the University of Oxford and has 100 technical papers, 14 patents and multiple books to his name, has been a crossword fan since he attended college 45 years ago.
But his obsession took off when he entered a tournament more than a decade ago and didn’t win.
“‘The other competitors were so much better than I was, and it annoyed me, so I thought ‘Well, I should write a program,’ so I started Dr.Fill,” Ginsberg says.
Organized by Shortz, the American Crossword Tournament is packed with people who know their way around words.
Dr.Fill made its debut at the competition in 2012. Despite high expectations, Dr.Fill only managed to place 141st out of 600 contestants. Dr.Fill never managed a top 10 finish until this year.
In part, that’s because crosswords didn’t attract the kind of richly funded efforts that took on — and eventually beat — the best humans at chess and go.
It’s also partly because crossword puzzles are unique. “In go and chess and checkers, the rules are very clear,” Ginsberg says. “Crosswords are very interesting.”
Crossword puzzles often rely on cryptic clues that require deep cultural knowledge and an extensive vocabulary, as well as the ability to find answers that best slide into each puzzle’s overlapping rows and columns.
“It’s a messy thing,” Shortz said. “It’s not purely logical like chess or even like Scrabble, where you have a word list and every word is worth so many points.”
Crossword puzzles often rely on cryptic clues that require deep cultural knowledge and an extensive vocabulary.
A Winning Combination
The game-changer? Help from the Natural Language Processing team. Inspired by his efforts, the team reached out to Ginsberg a month before the competition began.
It proved to be a triumphant combination.
The Berkeley team focused on understanding each puzzle’s often gnomic clues and finding potential answers. Klein’s team of three graduate students and two undergrads took the more than 6 million examples of crossword clues and answers that Ginsberg had collected and poured them into a sophisticated neural network.
Ginsberg’s software, refined over many years, then handled the task of ranking all the answers that fit the confines of each puzzle’s grid and fitting them in with overlapping letters from other answers — a classic constraint satisfaction problem.
While their systems relied on very different techniques, they both spoke the common language of probabilities. As a result, they snapped together almost perfectly.
“We quickly realized that we had very complementary pieces of the puzzle,” Klein said.
Together, their models parallel some of the ways people think, Klein says. Humans make decisions by either remembering what worked in the past or using a model to simulate of what might work in the future.
“I get excited when I see systems that do some of both,” Klein said.
The result of combining both approaches: Dr.Fill played almost perfectly.
The AI made just three errors during the tournament. Its biggest edge, however, was speed. It dispatched most of the competition’s puzzles in under a minute.
AI Supremacy Anything But Assured
But since, unlike chess or go, crossword puzzles are ever-changing, another such showing isn’t guaranteed.
“It’s very likely that the constructors will throw some curveballs,” Shortz said.
Ginsberg says he’s already working to improve Dr.Fill. “We’ll see who makes more progress.”
The result may be out to be even more engaging crossword puzzles than ever.
“It turns out that the things that are going to stump a computer are really creative,” Klein said.
It will help piece together a 3D map of the universe, probe subatomic interactions for green energy sources and much more.
Perlmutter, officially dedicated today at the National Energy Research Scientific Computing Center (NERSC), is a supercomputer that will deliver nearly four exaflops of AI performance for more than 7,000 researchers.
That makes Perlmutter the fastest system on the planet on the 16- and 32-bit mixed-precision math AI uses. And that performance doesn’t even include a second phase coming later this year to the system based at Lawrence Berkeley National Lab.
More than two dozen applications are getting ready to be among the first to ride the 6,159 NVIDIA A100 Tensor Core GPUs in Perlmutter, the largest A100-powered system in the world. They aim to advance science in astrophysics, climate science and more.
A 3D Map of the Universe
In one project, the supercomputer will help assemble the largest 3D map of the visible universe to date. It will process data from the Dark Energy Spectroscopic Instrument (DESI), a kind of cosmic camera that can capture as many as 5,000 galaxies in a single exposure.
Researchers need the speed of Perlmutter’s GPUs to capture dozens of exposures from one night to know where to point DESI the next night. Preparing a year’s worth of the data for publication would take weeks or months on prior systems, but Perlmutter should help them accomplish the task in as little as a few days.
“I’m really happy with the 20x speedups we’ve gotten on GPUs in our preparatory work,” said Rollin Thomas, a data architect at NERSC who’s helping researchers get their code ready for Perlmutter.
Perlmutter’s Persistence Pays Off
DESI’s map aims to shed light on dark energy, the mysterious physics behind the accelerating expansion of the universe. Dark energy was largely discovered through the 2011 Nobel Prize-winning work of Saul Perlmutter, a still-active astrophysicist at Berkeley Lab who will help dedicate the new supercomputer named for him.
“To me, Saul is an example of what people can do with the right combination of insatiable curiosity and a commitment to optimism,” said Thomas, who worked with Perlmutter on projects following up the Nobel-winning discovery.
Supercomputer Blends AI, HPC
A similar spirit fuels many projects that will run on NERSC’s new supercomputer. For example, work in materials science aims to discover atomic interactions that could point the way to better batteries and biofuels.
Traditional supercomputers can barely handle the math required to generate simulations of a few atoms over a few nanoseconds with programs such as Quantum Espresso. But by combining their highly accurate simulations with machine learning, scientists can study more atoms over longer stretches of time.
“In the past it was impossible to do fully atomistic simulations of big systems like battery interfaces, but now scientists plan to use Perlmutter to do just that,” said Brandon Cook, an applications performance specialist at NERSC who’s helping researchers launch such projects.
That’s where Tensor Cores in the A100 play a unique role. They accelerate both the double-precision floating point math for simulations and the mixed-precision calculations required for deep learning.
Similar work won NERSC recognition in November as a Gordon Bell finalist for its BerkeleyGW program using NVIDIA V100 GPUs. The extra muscle of the A100 promises to take such efforts to a new level, said Jack Deslippe, who led the project and oversees application performance at NERSC.
Software Helps Perlmutter Sing
Software is a strategic component of Perlmutter, too, said Deslippe, noting support for OpenMP and other popular programming models in the NVIDIA HPC SDK the system uses.
Separately, RAPIDS, open-source code for data science on GPUs, will speed the work of NERSC’s growing team of Python programmers. It proved its value in a project that analyzed all the network traffic on NERSC’s Cori supercomputer nearly 600x faster than prior efforts on CPUs.
“That convinced us RAPIDS will play a major part in accelerating scientific discovery through data,” said Thomas.
Coping with COVID’s Challenges
Despite the pandemic, Perlmutter is on schedule. But the team had to rethink critical steps like how it ran hackathons for researchers working from home on code for the system’s exascale-class applications.
Meanwhile, engineers from Hewlett Packard Enterprise helped assemble phase 1 of the system, collaborating with NERSC staff who upgraded their facility to accommodate the new system. “We greatly appreciate the work of those people onsite bringing the system up, especially under all the special COVID protocols,” said Thomas.
At the virtual launch event, NVIDIA CEO Jensen Huang congratulated the Berkeley Lab crew on its plans to advance science with the supercomputer.
“Perlmutter’s ability to fuse AI and high performance computing will lead to breakthroughs in a broad range of fields from materials science and quantum physics to climate projections, biological research and more,” Huang said.
On Time for AI Supercomputing
The virtual ribbon cutting today represents a very real milestone.
“AI for science is a growth area at the U.S. Department of Energy, where proof of concepts are moving into production use cases in areas like particle physics, materials science and bioenergy,” said Wahid Bhimji, acting lead for NERSC’s data and analytics services group.
“People are exploring larger and larger neural-network models and there’s a demand for access to more powerful resources, so Perlmutter with its A100 GPUs, all-flash file system and streaming data capabilities is well timed to meet this need for AI,” he added.